
Shares in Johnson & Johnson (NYSE:JNJ) have risen today after the medical giant posted first quarter earnings that topped expectations on Wall Street and lifted its EPS guidance for the full fiscal year.
The New Brunswick, N.J.-based company posted profits of approximately $3.75 billion, or $1.39 per share, on sales of $20 billion for the three months ended March 31, seeing the bottom line shrink 14.2% while sales grew 0.1% compared with the same period during the previous year.
Adjusted to exclude one-time items, earnings per share were $2.10, ahead of the $2.04 consensus on Wall Street where analysts expected to see sales of $19.6 billion, which the company also topped.
“Our strong first-quarter results reflect continued underlying operational sales and adjusted EPS growth. At the same time, we remain focused on investing in innovative technologies and platforms that will make a meaningful difference in the lives of patients around the world. I am proud of our global colleagues’ collective efforts to deliver on our long-term goals and our ability to create value for all of our stakeholders,” chair & CEO Alex Gorsky said in a press release.
Johnson & Johnson lifted its guidance for the remaining fiscal year, now expecting to see adjusted diluted EPS of between $8.53 and $8.63 up from previous guidance of between $8.50 and $8.65. The company maintains its sales expectation of between $80.4 billion and $81.2 billion.
Shares in Johnson & Johnson have risen 2.6% so far today, at $140.08 as of 9:47 a.m. EDT.
Earlier this month, Johnson & Johnson said that its its Ethicon biz completed its $3.4 billion acquisition of robotic surgery dev Auris Health and the $2.8 billion divestiture of its Advanced Sterilization Products to Fortive (NTSE:FTV).
from MassDevice http://bit.ly/2XhKmSy

 Glaukos
Glaukos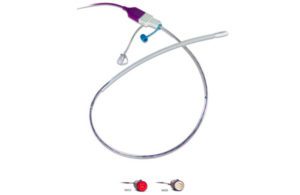

 The FDA last week granted pre-market approval to the Tack endovascular repair device developed by
The FDA last week granted pre-market approval to the Tack endovascular repair device developed by 
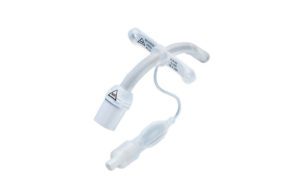






 The FDA said today that new data show that the contamination rate for “high-concern organisms” in duodenoscopes is nearly twice as high as previously thought and warned the devices three major manufacturers to get their required post-market studies in gear.
The FDA said today that new data show that the contamination rate for “high-concern organisms” in duodenoscopes is nearly twice as high as previously thought and warned the devices three major manufacturers to get their required post-market studies in gear.

 A state appeals court in Pennsylvania yesterday upheld the plaintiff’s $13.7 million win in a product liability lawsuit brought over one of
A state appeals court in Pennsylvania yesterday upheld the plaintiff’s $13.7 million win in a product liability lawsuit brought over one of 






 Shares in
Shares in 

 A quartet of U.S. representatives today introduced a companion bill to a Senate measure that would do away with the medical device tax altogether.
A quartet of U.S. representatives today introduced a companion bill to a Senate measure that would do away with the medical device tax altogether.

 The FDA today warned consumers against using medical devices purported to assess head injuries, saying tht only five companies are cleared to market such devices in the U.S. and that they should only be used by physicians.
The FDA today warned consumers against using medical devices purported to assess head injuries, saying tht only five companies are cleared to market such devices in the U.S. and that they should only be used by physicians.